Building a Protein Variant Classifier with ESM2 and Multi-GPU Training
Introduction
In the field of clinical genomics, accurately predicting whether a specific genetic variant is pathogenic (disease-causing) or benign is a critical challenge. Recently, I worked on a project to develop a deep learning model that classifies protein variants as either Gain-of-Function (GOF) or Loss-of-Function (LOF) using ESM2 (Evolutionary Scale Modeling), a state-of-the-art protein language model.
Challenge 1: Metric Selection for Clinical Use
Before diving into the model, I had to evaluate existing pathogenicity predictors. The dataset contains 107 patients, each with multiple variants where only a few are pathogenic (LABEL=1).
The Problem: Class Imbalance
The data is highly imbalanced—most variants are benign (LABEL=0), only a few are pathogenic (LABEL=1). This makes metric selection critical.
| Metric | Formula | Problem with Imbalanced Data |
|---|---|---|
| Accuracy | (TP+TN) / Total | Predicting all as benign gives high accuracy |
| AUROC | Area under TPR-FPR curve | Can look good even with poor precision |
Why AUROC Alone Is Not Enough
AUROC measures discrimination across all thresholds. A model with AUROC=0.94 sounds great, but:
- At what threshold does it achieve good Precision and Recall?
- In clinical diagnostics, False Negatives are dangerous (missing a pathogenic variant)
Metrics for Clinical Pathogenicity Prediction
For this problem, I focused on both classification metrics and ranking metrics:
Classification Metrics (Binary)
| Metric | Formula | Clinical Importance |
|---|---|---|
| Recall (Sensitivity) | $\frac{TP}{TP + FN}$ | Must be high: we cannot miss pathogenic variants |
| Precision (PPV) | $\frac{TP}{TP + FP}$ | Reduces unnecessary follow-up tests |
| F1 Score | $\frac{2 \times Precision \times Recall}{Precision + Recall}$ | Balances both for imbalanced data |
Ranking Metric (Patient-Centric)
Since each patient has multiple variants and we want the pathogenic variant to be ranked high:
| Metric | Formula | Clinical Importance |
|---|---|---|
| Top-K Recall | $\frac{\text{# patients with causal variant in top K}}{\text{# total patients}}$ | Measures how often the pathogenic variant appears in the top K predictions |
The formal definition:
\[\text{Top-K Recall} = \frac{1}{N} \sum_{i=1}^{N} \mathbb{1}[\text{rank}(v_i) \leq K]\]Where:
- $N$ = number of patients
- $v_i$ = the pathogenic variant for patient $i$
- $\text{rank}(v_i)$ = position when variants are sorted by prediction score (descending)
- $\mathbb{1}[\cdot]$ = indicator function (1 if true, 0 if false)
Why Recall Is Critical
In medical diagnostics, a False Negative (predicting benign when actually pathogenic) means:
- Patient doesn’t receive treatment
- Disease progresses undetected
Therefore, Recall must be prioritized, even at the cost of some False Positives.
Evaluation Framework
I evaluated each predictor (A, B, C) with both classification and ranking metrics:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
from sklearn.metrics import precision_recall_curve, roc_auc_score
import numpy as np
def evaluate_predictor(y_true: np.ndarray, y_scores: np.ndarray) -> dict:
"""Evaluate predictor with classification metrics."""
auroc = roc_auc_score(y_true, y_scores)
precisions, recalls, thresholds = precision_recall_curve(y_true, y_scores)
f1_scores = 2 * (precisions * recalls) / (precisions + recalls + 1e-8)
best_idx = np.argmax(f1_scores)
return {
"auroc": auroc,
"best_f1": f1_scores[best_idx],
"recall_at_best_f1": recalls[best_idx],
"precision_at_best_f1": precisions[best_idx],
}
# end def
def compute_top_k_recall(df: pd.DataFrame, score_col: str, k: int) -> float:
"""Compute Top-K Recall per patient."""
hits = 0
for patient_id, group in df.groupby("Patient_ID"):
sorted_group = group.sort_values(score_col, ascending=False)
top_k_labels = sorted_group.head(k)["LABEL"].values
if 1 in top_k_labels:
hits += 1
# end if
# end for
return hits / df["Patient_ID"].nunique()
# end def
Results
Classification Metrics
| Predictor | AUROC | Best F1 | Recall @ Best F1 | Precision @ Best F1 |
|---|---|---|---|---|
| A | 0.94 | 0.42 | 0.65 | 0.31 |
| B | 0.88 | 0.58 | 0.82 | 0.45 |
| C | 0.91 | 0.51 | 0.71 | 0.40 |
Ranking Metrics (Patient-Centric)
| Predictor | Top-1 Recall | Top-5 Recall |
|---|---|---|
| A | 12% | 35% |
| B | 24% | 52% |
| C | 18% | 41% |
Key Findings:
- Predictor A had the highest AUROC but the worst F1, Recall, and Top-K metrics
- Predictor B achieved 82% Recall and 52% Top-5 Recall—meaning it catches more pathogenic variants both in classification and ranking
Decision: For clinical use, Predictor B is preferred because:
- Highest Recall (minimizes missed pathogenic variants)
- Best F1 Score (balanced performance on imbalanced data)
- Best Top-5 Recall (pathogenic variant is in top 5 for 52% of patients)
Lesson: In medical AI with class imbalance, evaluate using multiple metrics that reflect clinical consequences—not just AUROC.
Challenge 2: Modeling Protein Variants with ESM2
The core task was to classify variants using esm2_t33_650M_UR50D.
Existing vs. Proposed Approach
A standard approach in this domain often involves feeding the mutant sequence directly into the model to predict its property.
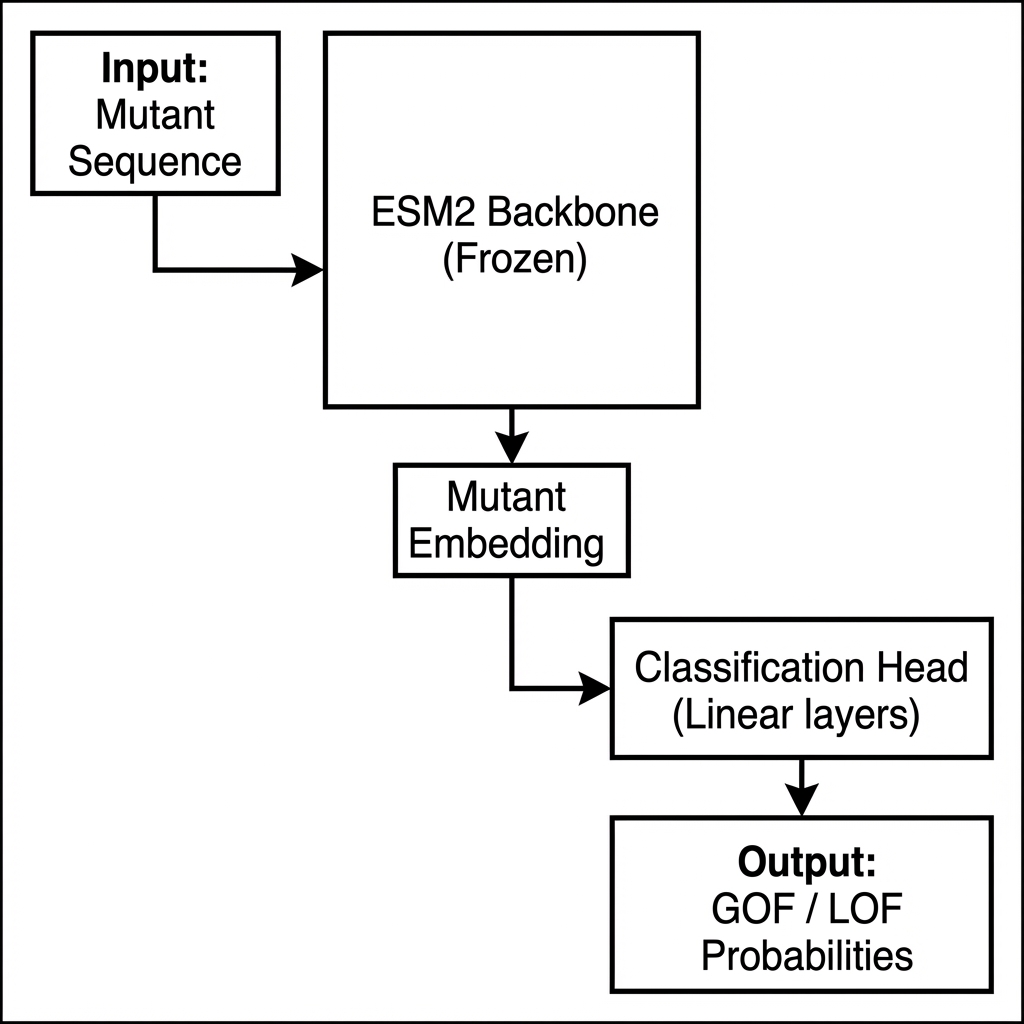 Figure 1: Standard Baseline Approach. The model only sees the mutant sequence, making it difficult to learn the specific impact of the mutation relative to the wild-type.
Figure 1: Standard Baseline Approach. The model only sees the mutant sequence, making it difficult to learn the specific impact of the mutation relative to the wild-type.
However, simply feeding the mutant sequence isn’t enough. The model needs to understand what changed. I designed the input to explicitly capture the difference:
1
Input = Concat(E_wt, E_mut, E_mut - E_wt)
- E_wt: Embedding of the Wild-Type sequence
- E_mut: Embedding of the Mutant sequence
- Difference: The vector representing the direction of change (Mutant - Wild-Type)
This “Difference Vector” proved crucial for distinguishing between LOF (function loss) and GOF (function gain).
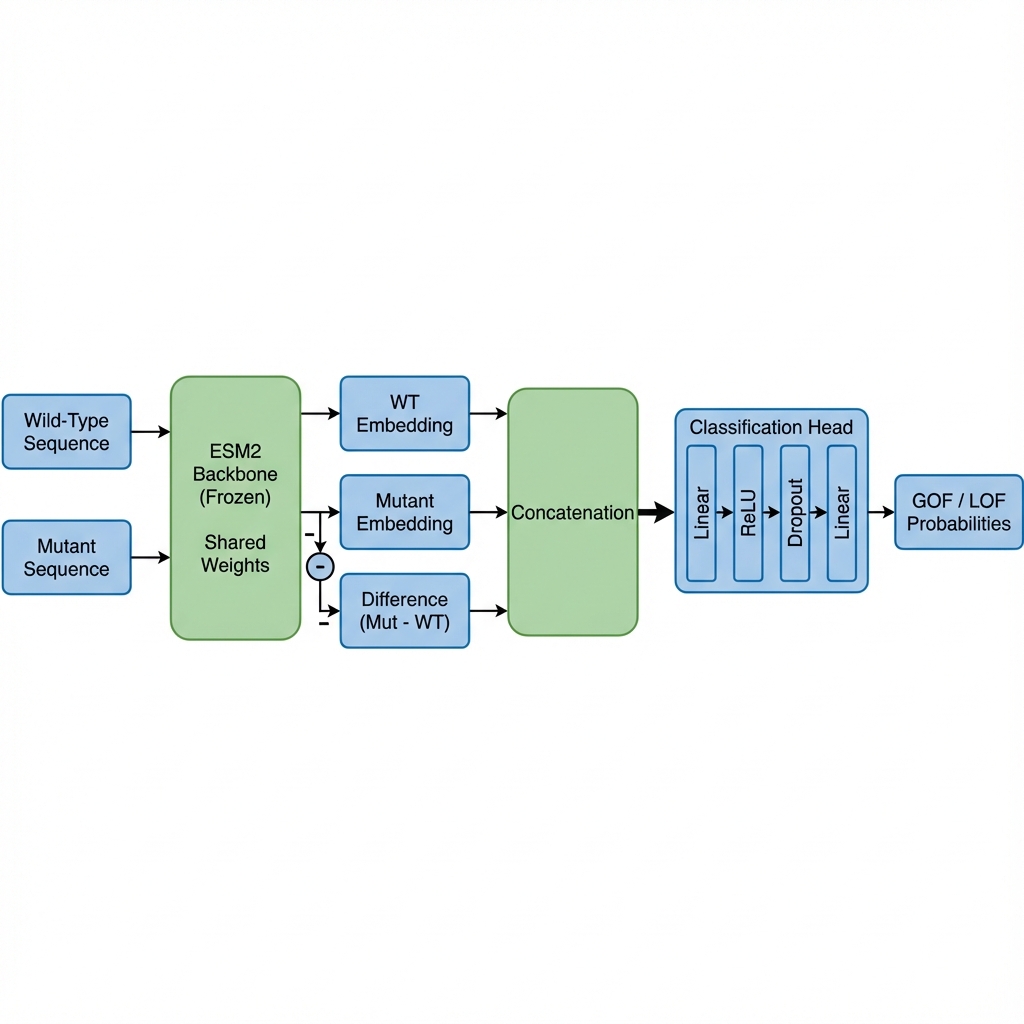 Figure 2: Our Proposed Architecture. By explicitly feeding the difference vector (Mutant - WT), the model can directly focus on the functional shift caused by the variant.
Figure 2: Our Proposed Architecture. By explicitly feeding the difference vector (Mutant - WT), the model can directly focus on the functional shift caused by the variant.
Code Snippet: Model Architecture
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
class ESM2VariantClassifier(nn.Module):
def __init__(self, model_name="facebook/esm2_t33_650M_UR50D"):
super().__init__()
self.esm = EsmModel.from_pretrained(model_name)
# Freeze backbone for efficiency
for param in self.esm.parameters():
param.requires_grad = False
hidden_size = self.esm.config.hidden_size
self.classifier = nn.Sequential(
nn.Linear(hidden_size * 3, 512), # 3x input size due to concatenation
nn.ReLU(),
nn.Dropout(0.3),
nn.Linear(512, 2)
)
def forward(self, wt_ids, wt_mask, mut_ids, mut_mask):
wt_out = self.esm(input_ids=wt_ids, attention_mask=wt_mask)
mut_out = self.esm(input_ids=mut_ids, attention_mask=mut_mask)
wt_cls = wt_out.last_hidden_state[:, 0, :]
mut_cls = mut_out.last_hidden_state[:, 0, :]
diff = mut_cls - wt_cls
combined = torch.cat((wt_cls, mut_cls, diff), dim=1)
return self.classifier(combined)
Challenge 3: Extreme Class Imbalance
The dataset had a 9:1 imbalance (90% LOF, 10% GOF). A standard model would simply predict “LOF” for everything and achieve 90% accuracy, which is useless.
Solution: Weighted Loss
I used CrossEntropyLoss with class weights inversely proportional to the class frequencies.
1
2
3
# LOF (0): 90%, GOF (1): 10%
class_weights = torch.tensor([0.1, 0.9]).to(device)
criterion = nn.CrossEntropyLoss(weight=class_weights)
This forces the model to pay 9x more attention to the minority GOF class, preventing it from being ignored.
Challenge 4: Distributed Training on A100s
To utilize 4x NVIDIA A100 GPUs, I used PyTorch’s DistributedDataParallel (DDP).
Key implementation details:
DistributedSampler: Ensures each GPU gets a different slice of data.init_process_group: Sets up communication between GPUs.torchrun: The launcher utility to manage processes.
One interesting hurdle was verifying DDP logic on a single local GPU. I learned that you can use the gloo backend (CPU-based) with torchrun --nproc_per_node=1 to simulate the distributed environment locally before deploying to the expensive cluster.
1
2
# Local verification command
torchrun --nproc_per_node=1 train_script.py --backend gloo
Conclusion
This project reinforced the importance of domain-specific feature engineering (Difference Vector) and robust engineering practices (DDP, Weighted Loss) when working with biological data. By combining pre-trained PLMs with thoughtful architecture, we can build powerful tools for genomic analysis.